El
cromosoma 22 es, tras el 23, el cromosoma más pequeño del que disponemos,
abarcando 51 millones de pares de bases (pb) y constituyendo únicamente entre
el 1,5 y el 2% del total de nuestro DNA y 545 genes y 134 seudogenes de los 20000-25000 que tiene
nuestro genoma.
Sin
embargo, tiene una cierta importancia histórica debido a que, como mencionamos en la introducción, fue el primer
cromosoma completamente secuenciado del Proyecto Genoma Humano.
 |
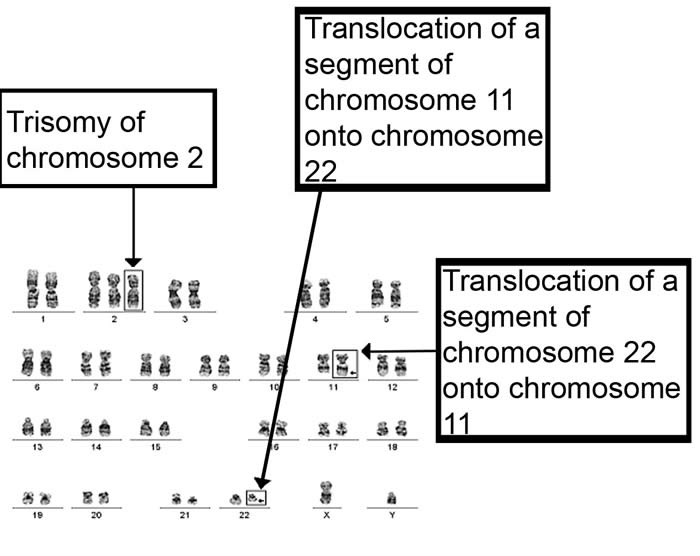
| Cariotipo de paciente con trisomía en el cromosoma 22 |
Si
bien existe la posibilidad de que haya una trisomía completa (que normalmente
causa la muerte del individuo durante el primer trimestre del embarazo), nos
centraremos más en el caso de que se dé un fenómeno de mosaicismo, es decir,
que sólo las células de algunos tejidos presenten dicha trisomía. Este tipo de
trisomía dará lugar a una serie de patologías relacionadas con los genes que
presenta dicho cromosoma:
 |
| Hipospadia |
-Síndrome
de Opitz G/BBB de tipo II: Esta patología es una enfermedad autosómica dominante que está relacionada
con el gen localizado en la posición 22q11.2 que cursa con hipotelorismo,
dificultad para tragar, defectos genitourinarios, especialmente hipospadias en
hombres and retraso mental y en el desarrollo y defectos cardiacos congénitos.
Normalmente, en los cromosomas 22 de los individuos con este síndrome
encontramos un anillo.
H
emos
encontrado estos síntomas en el caso [1].
-Síndrome
velocardiofacial: Otra de las patologías más comunes en los pacientes con trisomía 22 es
este síndrome relacionado con un gen también localizado en la región 22q11.21
que causa defectos cardiacos, dificultades en el aprendizaje o hipotonía
faríngea.
Estos
síntomas aparecen en el caso [2].
Por
tanto, teniendo en cuenta todas estas enfermedades, podemos establecer una
importante correlación entre la región 22q11 y las patologías asociadas a la
trisomía del cromosoma 22 por lo que todas estas enfermedades se agrupan bajo
el título de síndrome de deleción de 22q11.
 |
| Pacientes con síndrome de Emanuel |
-Síndrome de Emanuel: El síndrome de Emanuel está íntimamente relacionado con la trisomía en
el cromosoma 22 debida a la presencia de un cromosoma supernumerario que
consiste en fragmentos del cromosoma 11 y del cromosoma 22. Este nuevo cromosoma
se conoce como cromosoma derivado del 22, der (22).
Las personas con este síndrome normalmente heredan el
cromosoma der (22) de un padre no afectado por la enfermedad que lleva una
reorganización entre cromosoma 11 y cromosoma 22 llamada translocación
balanceada en la que ni se gana ni se pierde material genético. En esta
translocación no se pierde material genético. Los individuos con el síndrome de
Emanuel heredarán una translocación desbalanceada.
Se puede relacionar también con un gen presente
en la región 22q11.2 y provoca trastornos en el paciente tales como las hernias
inguinales, mandíbulas anchas, estrabismo o estenosis de la aorta y la arteria
pulmonar.
Podemos
encontrar dichos síntomas en el caso [3]
Síndrome
del ojo de gato: Es una enfermedad relacionada con el gen situado en la posición 22q11.
Esta es una de las enfermedades más típicas relacionadas con la trisomía del
cromosoma 22 debida a la presencia de un cromosoma supernumerario o extra.Esta enfermedad cursa con cataratas, colobomas (orificios
en el iris), fusiones en las vértebras o retrasos en el desarrollo
neuropsicológico.
Estos síntomas aparecen en el caso número [4]
 -Cromosoma
Filadelfia:
El cromosoma filadelfia es un cromosoma surgido de la translocación cromosómica
t (9; 22) (q34; q11) creando un nuevo gen híbrido denominado BCR/ABL que
codifica para una isoforma de la tirosin kinasa. Este nuevo cromosoma suele
estar relacionado con un tipo de leucemia denominado leucemia mieloide crónica
que está relacionado con un desorden en la citogenética de células madre
pluripotenciales lo que lleva a una afectación de las células de las estirpes
mieloide, eritroide, megacariocítica y linfoide (entre los linfocitos, los B
suelen ser los más afectados. Asimismo, un 10% de los pacientes con leucemia
linfocítica aguda presentan también esta translocación.
-Cromosoma
Filadelfia:
El cromosoma filadelfia es un cromosoma surgido de la translocación cromosómica
t (9; 22) (q34; q11) creando un nuevo gen híbrido denominado BCR/ABL que
codifica para una isoforma de la tirosin kinasa. Este nuevo cromosoma suele
estar relacionado con un tipo de leucemia denominado leucemia mieloide crónica
que está relacionado con un desorden en la citogenética de células madre
pluripotenciales lo que lleva a una afectación de las células de las estirpes
mieloide, eritroide, megacariocítica y linfoide (entre los linfocitos, los B
suelen ser los más afectados. Asimismo, un 10% de los pacientes con leucemia
linfocítica aguda presentan también esta translocación.
Un
ejemplo de esta enfermedad es el paciente con el caso clínico [5]
Formación del cromosoma Filadelfia
Paúl José Hernández Velasco
Bibliografía de
casos clínicos
[1]: Heinrich T, Nanda I, Rehn M, Zollner U, Frieauff E, Wirbelauer J, Grimm T,Schmid M. Live-born trisomy 22: patient report and review. Mol Syndromol. 2013Jan;3(6):262-9. doi: 10.1159/000346189. Epub 2013 Jan 11. PubMed PMID: 23599696; PubMed Central PMCID: PMC3569106
[2]: Shprintzen RJ, Goldberg RB, Young D, Wolford L. The velo-cardio-facial syndrome:
a clinical and genetic analysis. Pediatrics. 1981 Feb;67(2):167-72. PubMed
PMID: 7243439.
[3]: Carter MT, St Pierre SA, Zackai EH, Emanuel BS, Boycott KM. Phenotypic delineation
of Emanuel syndrome (supernumerary derivative 22 syndrome): Clinical features
of 63 individuals. Am J Med Genet A. 2009 Aug;149A(8):1712-21. doi:10.1002/ajmg.a.32957.
PubMed
PMID: 19606488; PubMed Central PMCID: PMC2733334.
[4]: Ko JM, Kim JB, Pai KS, Yun JN, Park SJ. Partial tetrasomy of chromosome 22q11.1 resulting from a supernumerary isodicentric marker chromosome in a boy with cat-eye syndrome. J Korean Med Sci. 2010 Dec;25(12):1798-801. doi: 10.3346/jkms.2010.25.12.1798. Epub 2010 Nov 24. PubMed PMID: 21165297; PubMed Central PMCID: PMC2995236.
[5]: Macedo MS, Figueiredo AR, Ferreira NN, Barbosa IM, Furtado MJ, Correia NF, Gomes MP, Lume MR, Menéres MJ, Santos MM, Meireles MA. Bilateral proliferative retinopathy as the initial presentation of chronic myeloid leukemia. Middle East Afr J Ophthalmol. 2013 Oct-Dec;20(4):353-6. doi: 10.4103/0974-9233.120016. PubMed PMID: 24339689;PubMed Central PMCID: PMC3841957
Enlaces
para obtener más información sobre dichas enfermedades:
 El día 2 de Diciembre de 1999 la prestigiosa revista científica Nature publicaba un trabajo colaborativo que informaba a la comunidad científica de la secuenciación del cromosoma 22 del cariotipo humano.
El día 2 de Diciembre de 1999 la prestigiosa revista científica Nature publicaba un trabajo colaborativo que informaba a la comunidad científica de la secuenciación del cromosoma 22 del cariotipo humano.